Cet article fait partie d’une série de textes rédigés par les anciens lauréats du Prix du Jeune chercheur de la SQHA résumant les développements récents dans leur domaine d’expertise
CES MITOCHONDRIES QUI NOUS FONT VIVRE ET NOUS TUENT
Yan Burelle, professeur agrégé, Faculté de pharmacie Université de Montréal
On sait depuis longtemps que les mitochondries sont le poumon de nos cellules, grâce à leur fonction génératrice d’ATP qui les place au cœur du métabolisme cellulaire. En contrepartie, on ne fait que commencer à apprécier comment les dysfonctions mitochondriales peuvent être à l’origine d’un vaste répertoire de pathologies affectant à peu près tout les systèmes physiologiques de notre organisme. En fait, on réalise que la contribution des ces organelles est omniprésente en physiopathologie ! Les avancées récentes en biologie mitochondriale nous démontrent que l’explication va bien au delà de ce que nous imaginions, il y a encore quelques années.
Des points névralgiques de la réponse immunitaire innée
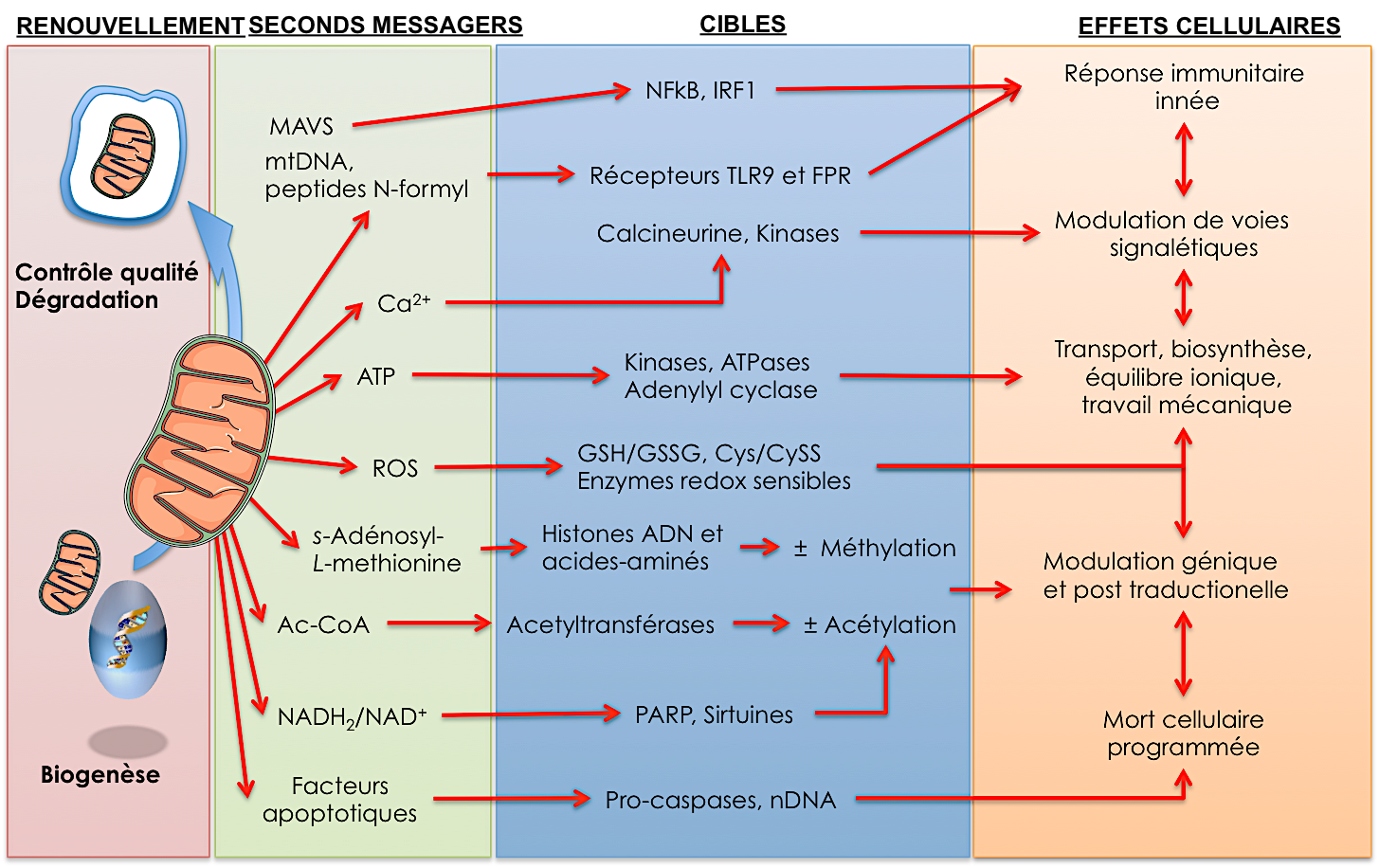
Les mitochondries apparaissent maintenant comme des senseurs et des médiateurs importants de la réponse immunitaire innée. En effet, les mitochondries agissent via les protéines MAVS (Mitochondria Antiviral Signaling proteins) situées sur leurs membranes externes, comme des plateformes de signalisation permettant d’orchestrer une réponse immunitaire adéquate suite à une infection virale 1. Des études récentes démontrent également que les mitochondries peuvent activer la réponse inflammatoire via la relâche de molécules alarmines. L’alarmine la mieux connue à ce jour est l’ADN mitochondrial lui-même, qui par sa structure plasmidique et ses motifs CpG non-méthylés, est reconnu comme de l’ADN bactérien par les récepteurs Toll-like-9. Les peptides N-formyls, produits par le clivage de certaines protéines mitochondriales, induisent également l’inflammation via l’activation des récepteurs N-formyl-like 2. De manière intéressante, il a été démontré que la relâche d’ADN mitochondrial dans le milieu intracellulaire des cardiomyocytes a la capacité de déclencher un processus inflammatoire local au cours de l’hypertrophie cardiaque pathologique3. Plusieurs études ont également rapporté la présence d’alarmines mitochondriales dans la circulation sanguine. À ce jour les études indiquent que ces alarmines contribuent vraisemblablement à la réponse inflammatoire systémique lors de pathologies induisant une mort cellulaire importante (ex : septicémie, infarctus, dommage hépatique, arthrite rhumatoïde) 2, mais leur rôle dans des processus inflammatoires chroniques de plus bas niveau – comme les dysfonctions et le remodelage cardiovasculaire dans l’hypertension par exemple – reste encore à être défini.
Les moteurs de l’adaptation cellulaire aux signaux énergétiques de l’environnement
Par ailleurs, on réalise maintenant que la bioénergétique mitochondriale constitue une interface essentielle permettant à la cellule de s’adapter à son environnement. En effet, les mitochondries, via la modulation des niveaux cellulaire d’ATP, de NAD+, d’acétyl-CoA et de méthionine, sont en prise directe avec les processus fondamentaux de régulation que sont la phosphorylation, l’acétylation et la méthylation des acides aminés et de l’ADN 4. Les mitochondries présentent également de grandes capacités à capter et relâcher le calcium, faisant d’elles des régulateurs de la signalisation calcique. Via plusieurs signaux métaboliques les mitochondries sont donc capables de moduler un grand nombre de cascades de signalisation et d’affecter l’expression des gènes nucléaires de leur cellule hôte par l’intermédiaire de mécanisme génétiques et épi-génétiques. En outre, plusieurs GPCR jusqu’à récemment orphelins s’avèrent être des récepteurs spécifiques de métabolites mitochondriaux relâchés dans le milieu extracellulaire: intermédiaires du cycle de Krebs, métabolites de la ?-oxydation, corps cétoniques, etc…5 Les mitochondries ont donc la capacité d’étendre leur rayon d’action signalétique bien au delà des frontières de leur cellule d’appartenance.
Certains facteurs de risque de maladie agiraient en perturbant ces mécanismes de signalisation, favorisant ainsi la propagation de dysfonctions mitochondriales et ultimement le développement de pathologies. Fait intéressant, des spécialistes de la génétique mitochondriale suggèrent que la susceptibilité variable des individus à développer un bon nombre de maladies chroniques est en partie reliée au génome des mitochondries elles-mêmes, notamment aux différents haplogroupes de l’ADN mitochondrial 6. Ces haplogroupes, exclusivement transmis par la mère au cours de l’évolution, sont caractérisés par des mutations ponctuelles ou des polymorphismes propres à l’ADN mitochondrial qui influencent vraisemblablement la signalisation rétrograde entre les mitochondries et le reste de la cellule, conférant ainsi une réponse différente aux facteurs de risques pour le développement de ces maladies. Notre génome n’explique pas tout. Il faut interroger celui de nos mitochondries !
Le renouvellement des mitochondries: une fontaine de jouvence
Finalement, une autre facette encore méconnue, mais qui suscite actuellement une grande curiosité, concerne les mécanismes de renouvellement des mitochondries. Ce renouvellement réfère en fait à deux processus simultanés qui globalement déterminent le cycle de vie et le fonctionnement de ces organelles. Le premier processus est la biogénèse, qui régule la synthèse de nouvelles mitochondries, et le second sont les processus de contrôle de qualité, qui régulent la dégradation des mitochondries dysfonctionnelles. Au plan de la biogenèse, plusieurs études démontrent qu’une perturbation de l’axe de signalisation régulé par PGC1?, un co-activateur transcriptionnel central dans ce processus, mine le potentiel des cellules à régénérer leur population mitochondriale et maintenir leur capacité anti-oxydantes 7. Pour ajouter au problème, il semble de plus en plus évident qu’une défaillance de certains processus de contrôle de qualité, puisse dans certains cas réduire la capacité des cellules à recycler leur mitochondries défectueuses, favorisant ainsi leur accumulation au cours du temps. Ce concept, encore nouveau, a récemment gagné en crédibilité suite au constat que plusieurs gènes (i.e. les gènes PARK) mutés chez des patients atteints de formes familiales précoces de Parkinson codent pour des protéines qui régulent la mitophagie (i.e. PINK1 et Parkin), un processus par lequel les mitochondries défectueuses sont séquestrées de manière sélective dans des autophagosomes et ensuite livrées au lysosomes pour leur dégradation 8. De manière intéressante, ce processus, qui n’est pas unique aux neurones dopaminergiques en cause dans la maladie de Parkinson, semble essentiel pour l’ensemble des tissus dotés de mitochondries 9. Il apparaît donc d’ores et déjà qu’une perte de capacité à renouveler adéquatement ses mitochondries soit une des causes fondamentales de l’accumulation de dysfonctions mitochondriales au cours du temps.
En conclusion, l’idée que les mitochondries qui nous permettent de vivre finissent un jour par nous tuer ne date pas d’hier. Aujourd’hui, nous commençons cependant à saisir un peu plus les mécanismes sous-jacents, qui sont loin de se limiter à la défaillance énergétique et au stress oxydant.
1. McWhirter, S. M., Tenoever, B. R. & Maniatis, T. Connecting mitochondria and innate immunity. Cell 122, 645–647 (2005).
2. Krysko, D. V. et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 32, 157–164 (2011).
3. Oka, T. et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 (2012).
4. Wallace, D. C. & Fan, W. Energetics, epigenetics, mitochondrial genetics. MITOCHONDRION 10, 12–31 (2010).
5. Blad, C. C., Tang, C. & Offermanns, S. G protein-coupled receptors for energy metabolites as new therapeutic targets. 1–17 (2012). doi:10.1038/nrd3777
6. Ballinger, S. W. Beyond retrograde and anterograde signalling: mitochondrial-nuclear interactions as a means for evolutionary adaptation and contemporary disease susceptibility. Biochem Soc Trans 41, 111–117 (2013).
7. Austin, S. & St-Pierre, J. PGC1a and mitochondrial metabolism–emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 125, 4963–4971 (2012).
8. Youle, R. J. & Narendra, D. P. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12, 9–14 (2011).
9. Chen, Y. & Dorn, G. W. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475 (2013).